蛋白质是生命的物质基础 , 每个蛋白质的氨基酸链扭曲、折叠、缠绕成复杂的结构 , 想要破解这种结构通常需要花很长的时间 , 甚至难以完成 。 截至目前 , 约有10万个蛋白质的结构已经用实验方法得到了解析 , 但这在已经测序的数10亿计的蛋白质中只占了很小一部分 。
但“看清”蛋白的结构和人类的很多疾病机理、药物研发等等息息相关 。 在蛋白质结构解析的几十年历史中 , X射线晶体学、核磁共振波谱学(NMR)、冷冻电镜(Cryo-SEM)技术纷纷发挥了巨大的贡献 , 但这些技术在科学界看来 , 都有着劳心劳力又价格高昂的缺点 。
如何简单地通过蛋白质的氨基酸序列来预测其形状?如何能解答这一问题 , 了解生命运作方式的将打开截然不同的一扇窗 。 这种设想提出的50多年后 , 谷歌旗下人工智能公司DeepMind在去年12月的国际蛋白质结构预测竞赛CASP上投下重磅 , 他们开发的基于神经网络的新模型AlphaFold2击败了其他选手 , 在预测准确性方面达到接近人类实验结果 , 让整个结构生物学界震惊 。

文章图片
北京时间7月15日 , DeepMind团队在顶级学术期刊《自然》(Nature)以“加快评审文章”(Accelerated Article Preview)形式在线发表了一篇题为“Highly accurate protein structure prediction with AlphaFold”的论文 , 全面详述了半年前造成轰动的这一模型 , 并首次对外分享开源代码 。 该论文于今年5月11日提交 , 7月12日被接收 。
DeepMind团队向澎湃新闻(www.thepaper.cn)采访人员提供了一份声明 , 公司创始人兼首席执行官Demis Hassabis在声明中表示 , 去年在CASP14大会上我们揭晓了一个可以将蛋白质3D结构预测精确到原子水平的全新AlphaFold系统 , 此后我们承诺会分享我们的方法 , 并为科学共同体提供广泛、免费的获取途径 。
“今天我们迈出了承诺的第一步 , 在《自然》期刊上分享AlphaFold的开源代码 , 并发表了系统的完整方法论 , 详尽细致说明AlphaFold是如何做到精确预测蛋白质3D结构的 。 作为一家致力于推动科学进步的公司 , 我们期待看到我们的方法将为科学界启发出什么其他新的研究方法 , 也期待很快能和大家分享更多我们的新进展 。 ”Hassabis表示 。

文章图片
值得一提的是 , 就在同一天 , 另一顶级期刊《科学》(Science)也在线发表了另一预测蛋白质结构的研究文章 , 题为“Accurate prediction of protein structures and interactions using a three-track neural network” 。
来自华盛顿大学、哈佛大学、德克萨斯大学西南医学中心等团队的研究人员开发了新的深度学习工具RoseTTAFold , 其拥有媲美AlphaFold2的蛋白质结构预测超高准确度 , 而且更快、所需计算机处理能力更低 。 同样 , 研究团队也对外分享了开源代码 。 该论文提交于6月7日 , 7月7日被接收 。
清华大学生命科学学院院长、高精尖中心执行主任王宏伟对澎湃新闻(www.thepaper.cn)采访人员表示 , “高质量结构预测的源代码开放对整个科学界尤其是结构生物学领域的促进作用必然是巨大的 。 ”他评价道 , 对于DeepMind这样一家商业公司来说 , “团队愿意向公众分享代码 , 是一个新型科研范式的突破 , 将整体上有利于人类更好地探索未知 。 ”
预测蛋白质结构 , 接近实验室测量
50多年前 , 科学家们就设想用计算机预测蛋白质结构 。 近年来 , 共同演化、接触图预测、深度机器学习等技术的引入 , 一些实验室的算法精度有了很大程度的提高 。
曾经开发出Alphago、战胜人类顶尖棋手的DeepMind团队是其中的佼佼者 , 其团队的强大和资源雄厚是一般实验室无法企及的 。 2020年12月1日 , 他们在生物领域展现出实力 , 在两年一度的权威蛋白质结构预测评估竞赛(CASP)中用AlphaFold2击败其他参赛团队 。
CASP是由马里兰大学John Moult教授等人于1994年组织 。 竞赛使用的是最新解决且尚未在蛋白质数据库(PDB)中存放或公开披露的结构 , 结构生物学家们利用X射线晶体学、核磁共振波谱学、冷冻电镜的方法 , 把这些蛋白质的结构解析出来 。 做蛋白质结构预测的团队则利用计算机程序来预测它们的结构 。 最后由独立的科学家团队则把计算机预测的模型和实验室的结构对照 , 分析不同计算机算法的预测结果 。 这是一种“双盲”测试 , 长期以来一直是评价结构预测准确性的金标准 。
去年的CASP14共有84个常规题目 , 其中有14题因为生物实验没给出确定结构等原因被取消或延缓 , 其他70个题目的单体和复合物蛋白质所含有的氨基酸个数从73到2180不等 。
19个国家的215个小组参加了CASP14 。 DeepMind公司的AlphaFold2预测的大部分结构达到了空前的准确度 , 不仅与实验方法不相上下 , 还远超解析新蛋白质结构的其他方法 。
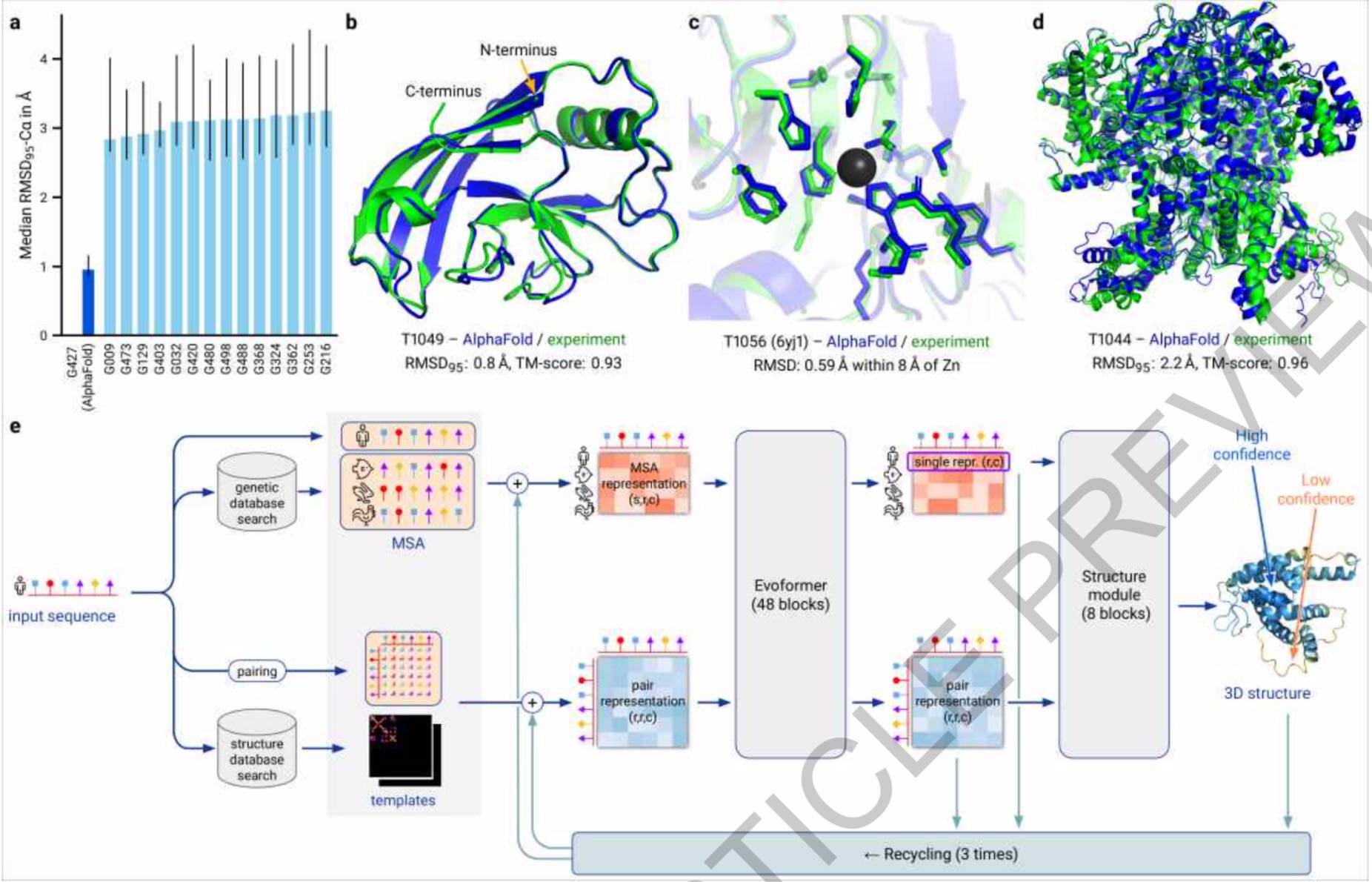
文章图片
将实验方法得到的蛋白质结构叠加在AlphaFold2的结构上 , 组成蛋白质主链骨架的叠加原子之间的距离中位数(95%的覆盖率)为0.96埃(0.096纳米) 。 成绩排第二的方法只能达到2.8埃的准确度 。
AlphaFold2的神经网络能在几分钟内预测出一个典型蛋白质的结构 , 还能预测较大蛋白质(比如一个含有2180个氨基酸、无同源结构的蛋白质)的结构 。 该模型能根据每个氨基酸对其预测可靠性进行精确预估 , 方便研究人员使用其预测结果 。
AlphaFold2最终被Moult评价道 , “在某种意义上 , 问题已经解决了” 。
值得一提的是 , 在最新发布的论文中 , DeepMind还简化了AlphaFold2 。 AlphaFold的首席研究员John Jumper说 , “这个网络需要几天的计算时间来生成CASP的一些蛋白质的结构 , 而开源版本的速度要快16倍 。 根据蛋白质的大小 , 它可以在几分钟到几小时内生成结构 。 ”
受AlphaFold2的启发 , 华盛顿大学医学院生物化学家、蛋白质设计研究所所长David Baker等人开发了RoseTTaFold 。 华盛顿大学医学院官网对该研究的介绍称 , 在高精度的蛋白质结构预测方面 , Baker等人“在很大程度上重现了DeepMind团队的表现 。 ”
相较于AlphaFold2只解决了单个蛋白质的结构 , RoseTTaFold不仅适用于简单的蛋白质 , 也适用于蛋白质复合物 。 据介绍 , RoseTTaFold利用深度学习技术 , 根据有限信息准确、快速地预测蛋白质结构 。 从结构上来看 , RoseTTAFold 是一个三轨(three-track)神经网络 , 它可以兼顾蛋白质序列的模式、氨基酸如何相互作用以及蛋白质可能的三维结构 。 在这种结构中 , 一维、二维、三维信息来回流动 , 使得网络能够集中推理蛋白质的化学部分与它的折叠结构 。
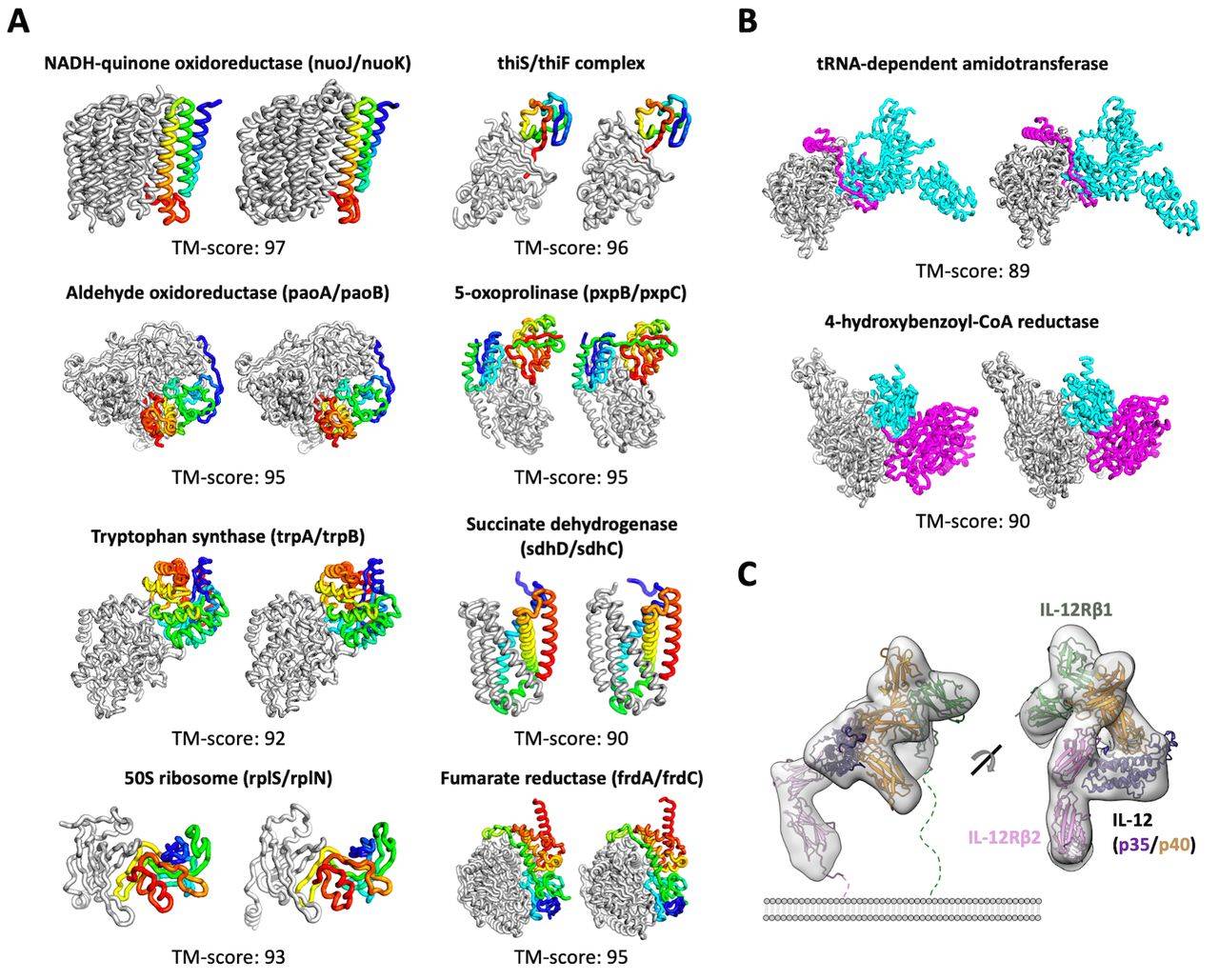
文章图片
巴塞尔大学的计算结构生物学家Torsten Schwede对《科学》杂志说 , 许多生物功能依赖于蛋白质之间的相互作用 。 “直接从序列信息中处理蛋白质-蛋白质复合物的能力使其对生物医学研究中的许多问题极具吸引力 。 ”
Baker同时坦言 , AlphaFold2的结构更加准确 。 但是根特大学的结构生物学家Savvas Savvides说 , Bake实验室的方法更好地捕捉到了“蛋白质结构的本质和特性” , 比如识别从蛋白质侧面伸出的原子串 , 这些特征是蛋白质之间相互作用的关键 。
纽约大学医学院的细胞和结构生物学家Gira Bhabha说 , 两种方法都很有效 。 她表示 , “DeepMind和Baker实验室的进展都是惊人的 , 将改变我们利用蛋白质结构预测推进生物学的方式 。 ”
开源代码 , 如何促进整个科学界?
相比于去年年底带来的震撼 , 这次外界更感兴趣的是上述两支团队开源代码这一动作 。
此前的6月中旬 , 在Baker实验室发布RoseTTAFold预印本三天之后 , DeepMind的Hassabis在推特上表示 , AlphaFold2的细节正在接受一份出版物的审查 , 公司将“为科学界提供广泛的免费访问” 。
而从6月1日开始 , Baker等人已经开始挑战他们的方法 , 让研究人员发送来他们最令人困惑的蛋白质序列 。 加州大学旧金山分校的结构生物物理学家David Agard的研究小组发送了一组没有已知类似蛋白质的氨基酸序列 , 几个小时内 , 他的团队就得到了一个蛋白质模型 , “这可能为我们节省了一年的工作 。 ”Agard说 。
除了免费提供RoseTTaFold的代码外 , Baker团队还建立了一个服务器 , 研究人员可以插入蛋白质序列并得到预测的结构 。 贝克说 , 自从上个月推出以来 , 该服务器已经预测了大约500人提交的5000多种蛋白质的结构 。
不过 , 上述两支团队的源代码都是免费的 , 但也有观点认为 , 对于没有技术专长的研究人员来说 , 它可能还不是特别有用 。 不过 , DeepMind的科学人工智能负责人Pushmeet Kohli表示 , DeepMind已经与一些选定的研究人员和组织合作 , 以预测特定的目标 , 其中包括总部位于瑞士日内瓦的非营利组织“Drugs for ignored Diseases” 。 “在这个领域 , 我们还有很多想做的事情 。 ”
Hassabis提到 , 去年在CASP14大会上我们揭晓了一个可以将蛋白质3D结构预测精确到原子水平的全新AlphaFold系统 , 此后我们承诺会分享我们的方法 , 并为科学共同体提供广泛、免费的获取途径 。 “今天我们迈出了承诺的第一步 , 在《自然》期刊上分享AlphaFold的开源代码 , 并发表了系统的完整方法论 , 详尽细致说明AlphaFold是如何做到精确预测蛋白质3D结构的 。 作为一家致力于推动科学进步的公司 , 我们期待看到我们的方法将为科学界启发出什么其他新的研究方法 , 也期待很快能和大家分享更多我们的新进展 。 ”
DeepMind团队认为 , 这一精准的预测算法可以让蛋白质结构解析技术跟上基因组革命的发展步伐 。
Baker团队也提到 , “我们希望这个新工具将继续造福整个研究界 。 ”
中国科学院合肥物质科学研究院强磁场科学中心研究员谢灿对澎湃新闻(www.thepaper.cn)采访人员表示 , “总的来说 , 对学术界来肯定是好事 , 肯定会促进结构生物学和相关领域的发展 。 在承认学术贡献的基础上的开放和共享 , 本来就应该是学术研究最基本的要求 。 ”
结构生物学是谢灿的“老本行” , “我当年花了8年的时间去解析一个蛋白的晶体结构 , 我能切身体会如果有一个精准预测蛋白结构的算法出现 , 对结构生物学家意味着什么 。 ”
但他认为 , 不必要担忧这些算法的出现会让结构生物学家失业 , 在技术迭代之下 , 结构生物学这些年受到的冲击太多了 , “而事实上 , 只不过是某一个领域某一个技术在某一个历史阶段更容易出工作出成绩 。 ”谢灿认为 , 无论再精准的预测 , 终究也只是预测 , “AlphaFold2不是实验 , 同样也需要实验去证实 。 ”
王宏伟在AlphaFold2刚出现之时也曾评价道 , 对于复杂的结构生物学问题 , 预测手段本身还不能号称完全解决了问题 。 实验结构生物学领域接下来需要做的一个事情是要拥抱变化 , 更好地与预测方法结合以及共同发展 。
相关论文链接:
https://www.nature.com/articles/s41586-021-03819-2
https://science.sciencemag.org/content/early/2021/07/14/science.abj8754
【科学界|蛋白质结构预测哪家强?两大国际团队同日在顶刊开源代码】(本文来自澎湃新闻 , 更多原创资讯请下载“澎湃新闻”APP)
推荐阅读







- 最新消息|汽车之家回应裁员传闻:系正常业务结构优化、涉及人员不到5%
- 载体|可储氢的“纳米巧克力”结构创建
- Tesla|新航拍视频显示特斯拉在Model Y上测试结构性电池组
- 旗舰|雷军:小米12采用新一代钴酸锂电池、三明治结构电路板,成本更高
- 人工智能|人工智能预测蛋白质结构
- 棉花|国金早报|宁波“清零”!深圳同一落地航班检出3例奥密克戎;英国日增新冠确诊首超12万例;南方电网股权结构调整
- 机械|努力深化人才培养供给侧结构性改革——南昌工程学院机器人现代产业学院成立
- 协同|中科合肥智慧农业协同创新研究院项目地下结构封顶
- 实验|AI预测蛋白质结构,正在加快新药研发进程
- 预测|AI预测蛋白质结构登上Science、Nature年度技术突破,潜力无穷